Note
Go to the end to download the full example code.
Ground state analysis for NiO¶
This example follows the Anderson impurity model for NiO XAS example and considers the same model. This time we show how to analyze the wavevectors in terms of a \(\alpha |d^8L^{10}> + \beta |d^9L^9> \gamma |d^{10}L^8>\) representation.
In doing this we will go through the exercise of building and diagonalizing the Hamiltonian in a way that hopefully clarifies how to analyze other properties of the model.
import edrixs
import numpy as np
import matplotlib.pyplot as plt
import scipy
import example_3_AIM_XAS
import importlib
_ = importlib.reload(example_3_AIM_XAS)
Hamiltonian¶
edrixs builds model Hamiltonians based on two fermion and four fermion terms.
The four fermion terms come from Coulomb interactions and will be
assigned to umat. All other interactions contribute to two fermion
terms in emat.
\[\begin{equation} \hat{H}_{i} = \sum_{\alpha,\beta} t_{\alpha,\beta} \hat{f}^{\dagger}_{\alpha} \hat{f}_{\beta} + \sum_{\alpha,\beta,\gamma,\delta} U_{\alpha,\beta,\gamma,\delta} \hat{f}^{\dagger}_{\alpha}\hat{f}^{\dagger}_{\beta} \hat{f}_{\gamma}\hat{f}_{\delta}, \end{equation}\]
Import parameters¶
Let’s get the parammeters we need from the
Anderson impurity model for NiO XAS example. We need to
consider ntot=20 spin-orbitals
for this problem.
Four fermion matrix¶
The Coulomb interactions in the \(d\) shell of this problem are described
by a \(10\times10\times10\times10\) matrix. We
need to specify a \(20\times20\times20\times 20\) matrix since we need to
include the full problem with ntot=20 spin-orbitals. The edrixs
convention is to put the \(d\) orbitals first, so we assign them to the
first \(10\times10\times10\times 10\) indices of the matrix. edrixs
creates this matrix in the complex harmmonic basis by default.
Two fermion matrix¶
Previously we made a \(10\times10\) two-fermion matrix describing the \(d\)-shell interactions. Keep in mind we did this in the real harmonic basis. We need to specify the two-fermion matrix for the full problem \(20\times20\) spin-orbitals in size.
The bath_level energies need to be applied to the diagonal of the
last \(10\times10\) region of the matrix.
The hyb terms mix the impurity and bath states and are therefore
applied to the off-diagonal terms of emat.
We now need to transform into the complex harmonic basis. We assign the two diagonal blocks of a \(20\times20\) matrix to the conjugate transpose of the transition matrix.
The spin exchange is built from the spin operators and the effective field is applied to the \(d\)-shell region of the matrix.
Build the Fock basis and Hamiltonain and Diagonalize¶
We create the fock basis and build the Hamiltonian using the full set of \(20\) spin orbitals, specifying that they are occuplied by \(18\) electrons. See the Exact diagonalization example for more details if needed. We also set the ground state to zero energy.
State analysis¶
Now we have all the eigenvectors in the Fock basis, we need to pick out the states that have 8, 9 and 10 \(d\)-electrons, respectively. The modulus squared of these coeffcients need to be added up to get \(\alpha\), \(\beta\), and \(\gamma\).
num_d_electrons = basis[:, :norb_d].sum(1)
alphas = np.sum(np.abs(v[num_d_electrons==8, :])**2, axis=0)
betas = np.sum(np.abs(v[num_d_electrons==9, :])**2, axis=0)
gammas = np.sum(np.abs(v[num_d_electrons==10, :])**2, axis=0)
The ground state is the first entry.
Ground state
alpha=0.825 beta=0.171 gamma=0.004
Plot¶
Let’s look how \(\alpha\), \(\beta\), and \(\gamma\) vary with energy.
fig, ax = plt.subplots()
ax.plot(e, alphas, label=r'$\alpha$ $d^8L^{10}$')
ax.plot(e, betas, label=r'$\beta$ $d^9L^{9}$')
ax.plot(e, gammas, label=r'$\gamma$ $d^{10}L^{8}$')
ax.set_xlabel('Energy (eV)')
ax.set_ylabel('Population')
ax.set_title('NiO')
ax.legend()
plt.show()
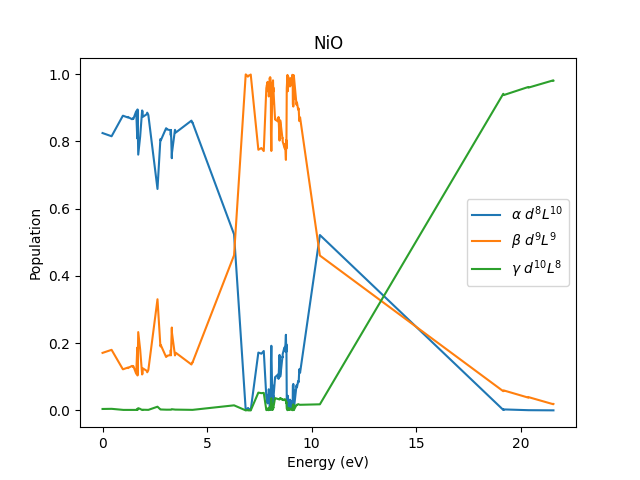
We see that the ligand states are mixed into the ground state, but the majority of the weight for the \(L^9\) and \(L^8\) states live at \(\Delta\) and \(2\Delta\). With a lot of additional structure from the other interactions.
Total running time of the script: (0 minutes 0.421 seconds)